INTRODUCTION
The TAGC consortium was founded in 2011 by 11 labs from the NY area and Israel, with the goal of generating a comprehensive catalog of genetic variation in the Ashkenazi Jewish population. In our first phase, we have generated high-coverage, whole genome sequences for 128 healthy individuals of Ashkenazi Jewish descent, as described in our paper in Nature Communications (2014). In Phase 2, we will sequence additional 200 healthy Ashkenazi individuals, in collaboration with the New York Genome Center.
UTILITY
Our dataset is the only publicly-available, reference sequencing data for the Ashkenazi Jewish population. As such, we expect it to be of utmost importance across the entire spectrum of genetic studies in this population. Specific applications include refinement of carrier screening panels, interpretation of clinical genomes, imputation of missing variants in sparsely genotyped samples, demographic inference, IBD mapping, and others. Our sequencing dataset is also expected to be useful for method development or comparative analysis in statistical and population genetics studies in other populations, in particular isolated ones.
For a more complete description of the project and the results of the data analysis, see here.
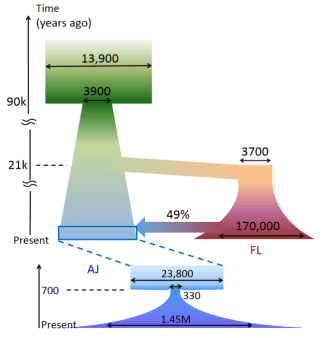
A reconstruction of the Ashkenazi Jewish and European demographic history using DNA sequence data. (AJ: Ashkenazi Jews; FL: Flemish from Belgium). The upper part shows the reconstruction of the ancient history of these populations, while the lower part illustrates the recent AJ history. Horizontal arrows correspond to effective population size. The wide arrow represents a migration event. The ancient reduction in population size on the left side likely corresponds to an Out-Of-Africa event and the formation of the Middle-Eastern population. This was followed by the divergence of Europeans, members of which later migrated into the Ashkenazi population. From Carmi et al., Nat. Commun., 2014.

The lengths of shared segment in Ashkenazi Jews (AJ). The observed pattern in AJ cannot be explained by a constant population size, but is very well fitted into a population undergoing a severe bottleneck (reduction in population size) followed by rapid expansion. From Carmi et al., Nat. Commun., 2014.
